Etc/생물정보 파일구조
생물정보 파일구조 - FASTA, FASTQ, BAM, SAM
gmta
2022. 1. 20. 15:03
반응형
FASTA
- 특정 분자의 서열을 나타내는데 사용합니다
- 주로 Genome의 각 chromosome 마다의 서열을 저장하는데 쓰입니다.
- 두 형태가 반복되는 형식의 구조입니다.
- 첫 부분은 아래에 나올 서열의 이름/ID 입니다.
- ' > ' (꺽쇠) 글자로 시작합니다.
- 다음에 이름에 해당하는 서열이 나오게 됩니다.
- 보통 서열은 굉장히 길며, 특정 유전자나 단백질, 염색체 등이 몇백~몇만 글자로 표현해야합니다.
- 그렇기 때문에 한줄로 나타내지 않고, 보통 60 글자로 나눠서 여러 줄에 걸쳐 표현합니다.

FASTQ
- NGS(Next Generation Sequencing data)의 결과를 저장하는 데 주로 사용됩니다.
- NGS 실험을 진행하면 결과로 cDNA library 서열을 읽어서 데이터로 얻을 수 있습니다. 즉, cDNA library 의 염기서열을 알 수 있습니다.
- 이 서열 하나를 'read' 라고 하며, fastq 파일은 여러 read 의 정보를 한 파일에 저장하게 됩니다.
- 이러한 파일은 직접 사용보다 reference genome 에 align 한 뒤에 활용됩니다.
- fastq 파일은 대부분의 연구에서 raw data, 즉 가공되지 않은 원본 파일로 여겨집니다.
- 파일은 네 줄이 한 단위입니다. cDNA library 정보가 네 줄에 나눠서 표현되어 있습니다.
- sequence ID: @ 로 시작하며, 해당 서열의 이름을 나타냅니다.
- sequence: 실제로 읽은 염기서열 정보입니다.
- description: '+' 글자로 시작하며 '+' 하나만 있기도 하고 sequence ID를 넣거나 설명을 넣는 부분입니다.
- quality: 각 염기서열이 얼마나 정확히 읽혔는지를 나타냅니다.

BAM
- BAM은 binary alignment map 이라는 형식입니다.
- fastq 파일을 reference genome에 align 했을 때 만들어지는 파일입니다.
- 각 cDNA library 조각이 reference genome의 어떤 부분에서 나왔는지에 대한 정보를 담는 것 입니다.
- fastq에서 각 read의 염기서열과 품질을 알 수 있다면, BAM 파일은 염기서열과 reference 에서의 위치정보를 알 수 있습니다.
- BAM 파일은 사람이 읽을 수 없는 파일입니다.
SAM
- BAM 파일과 SAM 파일은 동일한 정보를 가지고 있고, 서로 변환이 가능합니다.
- SAM 파일은 header 부분과 alignment 부분으로 이루어져 있습니다.
- header: 파일에 대한 설명을 주는 부분입니다. @로 시작하는 라인들입니다.
- alignment: 각 read에 대한 alignment 정보를 제공하는 부분입니다. 필수적으로 11개의 컬럼으로 이루어져 있고, 추가로 몇 개의 컬럼이 더 있을 수 있습니다.
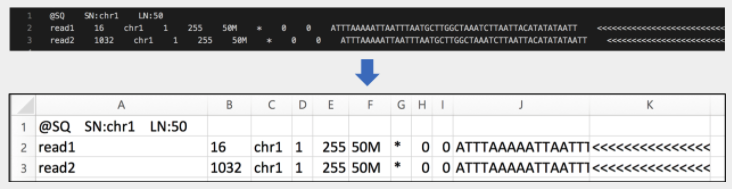
* 각 column 에 대한 설명
- QNAME: read 이름
- FLAG: 2진수로 된 read alignment 에 대한 설명
- RNAME: reference sequence의 이름
- POS: reference sequence 에서 align 된 위치
- MAPQ: mapping quality.(얼마나 정확히 align 되었는지 표시)
- CIGAR string: alignment 정보를 표현한 문자열. Match, Gap 등의 설명을 각 염기마다 표현합니다.
- RNEXT: 다음 read의 reference sequence 이름(paired end read에 대한 분석을 위해 사용됩니다.)
- PNEXT: 다음 read의 align 된 위치. 주로 paired end read에 대한 분석을 위해 사용됩니다.
- TLEN: Template length. paired-end read 둘의 left-end 부터 right-end까지의 길이를 의미합니다.
- SEQ: segment sequence(염기서열을 나타냅니다.)
- QUAL: Phread quality score입니다.